GTFermiSurface
GTFermiSurface[Hamiltonian,Fermi energy,list of bands,ndel, kbasis,clusterdata,BZPath]
calculates the Fermi surface corresponding to a Hamiltonian and Fermi energy if the Fermi surface contains parts from list of bands. The electronic structure is calculated in a cube at ndel points per spatial dimension. kbasis is the basis of the reciprocal lattice. clusterdata contains the data for the lattice construction. The path used in electronic structure calculations can be provided by BZPath.
Details
- The Mathematica function ListPlot3D is an appropriate tool to construct the Fermi surface as an isoenergy surface of the band structure. The band structure will be calculated in an interval of
 for all spatial dimensions in k-space, i.e. in a cube. In each direction the electronic structure is calculated at ndel points. kbasis, clusterdata and BZPath are used to construct the Brillouin zone.
for all spatial dimensions in k-space, i.e. in a cube. In each direction the electronic structure is calculated at ndel points. kbasis, clusterdata and BZPath are used to construct the Brillouin zone. - clusterdata defines the lattice vectors used for the construction of the Voronoi cell:
- clusterdata = {cut,smin,smax}
- cut is the maximum length of the lattice vectors. Lattice vectors between the shell numbers smin and smax are taken into account.
- The following options can be given:
-
GOBZ True Plots the Brillouin zone. GOBZPath False Plots the Brillouin zone path. GORegionFunction False Cuts away parts of the Fermi surface in adjacent Brillouin zones. GOVerbose False Controls the output of additional information. - See: W. Hergert, M. Geilhufe, Group Theory in Solid State Physics and Photonics. Problem Solving with Mathematica, chapter 9.7.1.
- Description of the XCrySDen software at http://www.xcrysden.org
Examplesopen all
Basic Examples (1)
| In[1]:= |
| In[2]:= |
Read the TB parameterset for Cu.

Read the predefined ![]() Hamiltonian for the fcc structure.
Hamiltonian for the fcc structure.
| In[4]:= |
The Hamiltonian for Cu is prepared by insertion of the parameter set.
| In[5]:= |
To see that all works correctly, the band structure is calculated. This is not necessary for the construction of the XCrySDen files.

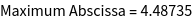

The exact position of the Fermi energy can be found from the DOS. We calculate the isoenergetic surface to E = 0.6 Ryd.